Blog Post
Corrective and Preventive Action (CAPA): Definition, Process, and Examples
CAPA is a critical process to dig into the root cause and avoid future recurrences. Learn what CAPA is and how it can benefit your operations.



Key Takeaways
CAPA programs most often fail at the follow-up, not the investigation. The effectiveness check gets dropped, the failure recurs, and the audit record documents the whole cycle.
Regulators assess corrective and preventive action separately, and the distinction matters at inspection. Filing corrective actions as preventive ones, or running a reactive-only program, is one of the most common bases for an FDA 21 CFR 820.100 observation.
Root cause analysis that stops at the visible cause produces a corrective action that closes on paper but not in practice. The failure mode stays in the asset. The CAPA reopens.
A CAPA program is only as good as the asset history behind it. When work orders, failure events, and maintenance records live in disconnected systems, the investigation starts with incomplete evidence, and the audit record reflects that.
If you close a CAPA without checking whether it worked, you've just done the paperwork, not the job. The asset goes back into service, and the same condition that caused the failure is still there. The next breakdown is already scheduled, you just don't know when.
CAPA programs fail at the same place every time: The investigation stops at what's visible, the action fixes the symptom, and nobody follows up because there's no system that forces them. The CAPA closes, the failure comes back, and the record shows the whole cycle, which is exactly what an inspector reads when they pull the file.
What Is Corrective and Preventive Action?
Corrective and preventive action (CAPA) is a structured quality management process for identifying, investigating, and eliminating the root causes of non-conformities and undesirable situations, whether those situations have already occurred or have the potential to occur. The acronym covers two distinct processes that trigger, operate, and are evaluated differently by regulatory bodies.
Running a reactive-only program that opens CAPAs in response to failures but never addresses potential risks satisfies the form requirement but not its intent. Regulators distinguish between the two.
Corrective action addresses failures that have already occurred. Preventive action handles conditions that could produce a failure before it does.
These two are frequently conflated, including in CAPA programs that pass casual audits but fail under scrutiny. However, the FDA CDRH defines them as three separate categories with separate purposes, and inspectors treat them that way.
|
Correction |
Corrective Action |
Preventive Action |
|
|---|---|---|---|
|
Definition |
Immediate fix that eliminates a detected nonconformity |
Action to eliminate the cause of a detected nonconformity and prevent recurrence |
Action to eliminate the cause of a potential nonconformity before it occurs |
|
Trigger |
A specific failure or defect has occurred |
A failure, audit finding, or repeated deviation has occurred |
Data, trends, risk assessment, or near-miss; no failure has occurred yet |
|
Goal |
Restore function |
Remove the root cause so the failure mode can’t recur |
Address conditions that could produce a failure before it does |
|
Maintenance Example |
Replace a failed pump bearing; return equipment to service |
Investigate why the bearing failed prematurely; adjust PM interval or lubrication spec to eliminate the cause |
Work order trend shows increasing bearing failures across similar assets; inspect and service the fourth asset before it fails |
Regulatory Framework
CAPA requirements appear across several major quality and regulatory frameworks, each with a slightly different scope and emphasis:
FDA 21 CFR 820.100 requires medical device manufacturers to establish and maintain documented procedures for both corrective and preventive action. The regulation specifies data analysis, root cause investigation, action implementation, effectiveness verification, and management review as required elements.
Both corrective and preventive actions must be verified or validated to confirm they’re effective and don’t introduce new problems.
ISO 9001:2015 replaced the explicit preventive action clause from its 2008 predecessor with risk-based thinking distributed throughout the quality management system. This shift is sometimes read as a signal that preventive action matters less. The opposite is true. The 2015 revision treats prevention as a system-level posture built into planning and operational review, rather than an administrative task triggered by a specific form.
The ASQ notes that quality professionals frequently express confusion on this point: A corrective action deals with a nonconformity that has occurred, while a preventive action addresses the potential for one to occur.
ISO 13485 for medical device quality management systems retains explicit requirements for corrective and preventive action as separately auditable processes, regardless of the ISO 9001:2015 shift.
In GMP environments, such as pharmaceutical manufacturing, food safety, and aerospace, CAPA is embedded in the quality system as a compliance requirement rather than an improvement option. Maintenance records are part of that quality system and are subject to audit.
What Makes Corrective Action Work and Where It Breaks Down
The investigation is where corrective action either holds or doesn't. A 5 Whys analysis that stops at operator error hasn't found the root cause, just a person to blame.
The next question is why the operator was in a position to make that error: the training gap, the procedure that was ambiguous, the scheduling condition that will produce the same outcome with a different operator next month.
Corrective action also breaks down at closure. The verification standard needs to be defined before the action closes, not when someone reviews the file six months later. For a recurring equipment failure, two complete PM cycles without recurrence is a reasonable baseline.
Without a defined criterion and a follow-up date built into the workflow, the verification never happens. That's where the loop restarts.
What Makes Preventive Action Work and Why Most Programs Skip It
Preventive action fails at the trigger. Teams don’t initiate it because nothing reveals the signal. A work order trend showing increasing pump seal failures across three similar assets should prompt an inspection of the fourth before it goes down. It rarely does though, because that pattern only becomes visible if someone is actively looking for it.
In a reactive operation, nobody is. The signal exists in the data only and never becomes a work order.
The documentation requirement mirrors corrective action: Record the signal, the potential failure mode, the change made, and how effectiveness will be confirmed. The difference is, there’s no failure event forcing the process open. That's the gap most programs never close.
The CAPA Process: Steps and Requirements
A functioning CAPA process follows a documented sequence, each step with a specific output. When steps are compressed or dropped, the gaps appear in the record and become the basis for audit observations.


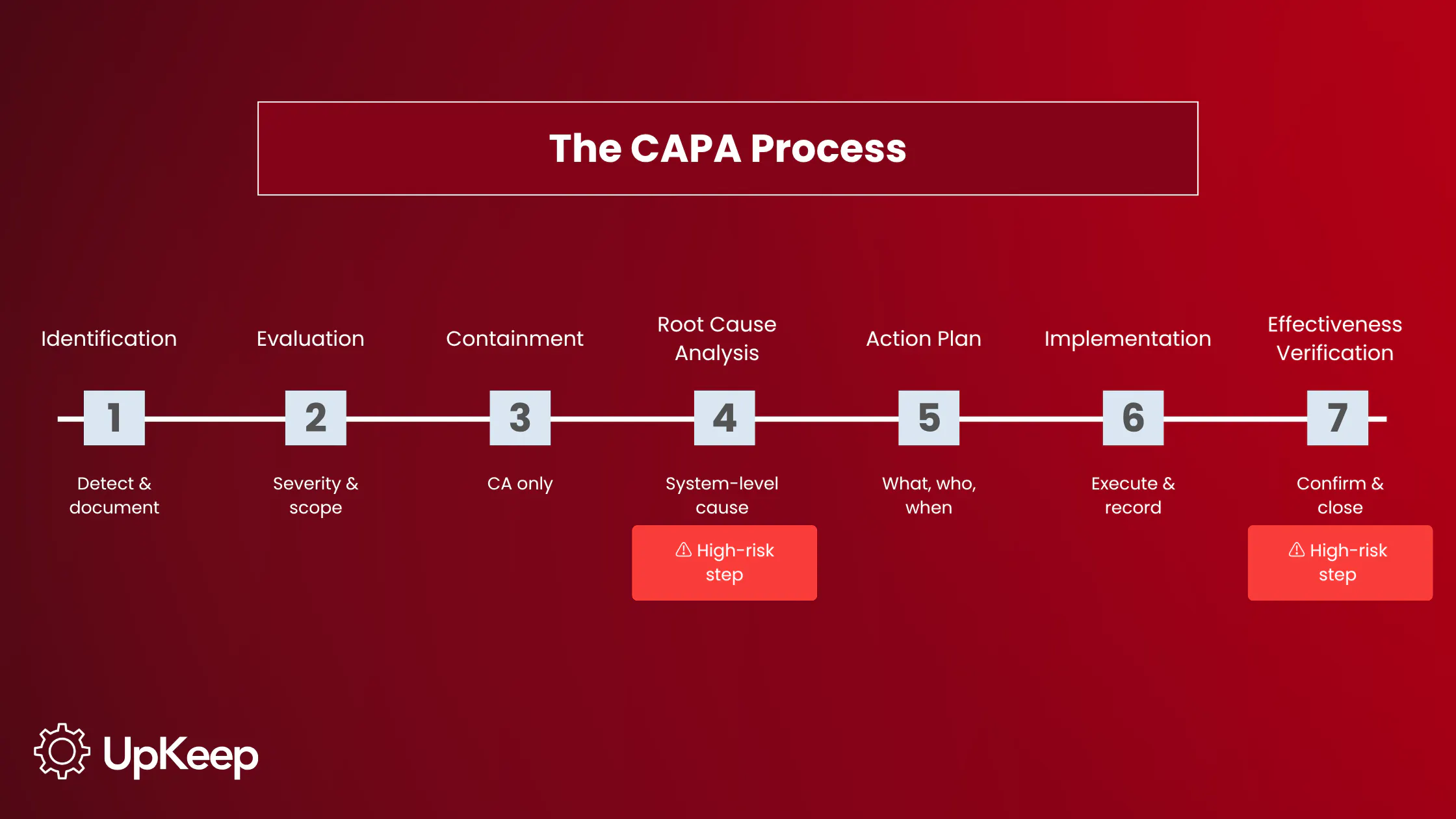
Identification
Detect the non-conformity, complaint, deviation, or risk signal. Document it with enough specificity to support investigation.
A problem statement reading "equipment malfunction" doesn’t support a root cause investigation. "Heat exchanger fouling caused unplanned downtime on Line 3 on three occasions in the past 60 days" does.
Evaluation
Assess severity and scope. Is the problem isolated or systemic? Severity determines how quickly the investigation proceeds and how rigorously the corrective action must be validated.
Containment (corrective action only)
Take immediate action to stop the problem from spreading: quarantine a batch, take a line offline, suspend a process.
Containment isn’t corrective action though. A CAPA record presenting containment as the corrective action hasn’t addressed the root cause.
Root Cause Analysis
This is where CAPA programs most often break down. The root cause is the system-level condition that produced the failure, not the failure itself or the proximate trigger.
An investigation that identifies "heat exchanger fouling" as the root cause of a production line shutdown has identified a symptom. The root cause is why fouling accelerated, whether that’s a PM interval set to OEM defaults that assumed clean process fluid, a process change that increased particulate load and was never reflected in the service schedule, or a work order closed without confirmation that the cleaning procedure was completed.
Action Plan Development
Define what changes, who owns it, and when it’ll be completed. Actions must address the root cause.
Retraining the technician who missed the lubrication step is correcting a symptom if the root cause is a PM interval set too wide. Retraining may be part of the action, but it’s insufficient on its own if the scheduling condition remains unchanged.
Implementation
Execute the plan and document every step. In regulated environments, the implementation record is part of the audit trail. An action taken but not documented is, from a regulatory standpoint, an action not taken.
Effectiveness Verification
After sufficient time has passed, confirm the failure mode hasn’t recurred and that the corrective action didn’t introduce new problems.
Define the verification criterion before closing the action, not after. This is the most commonly cited gap in FDA 483 observations related to CAPA programs.
Corrective vs. Preventive Action: How to Tell the Difference
The most common mistake is filing a corrective action as preventive because it prevents the problem from happening again. That's not the distinction.
Preventing the recurrence of something that’s already happened is corrective. Preventing the occurrence of something that hasn't happened yet is preventive. The trigger determines the category, not the intent.
Say a pump bearing fails, and the investigation finds the PM interval was set to OEM defaults and never updated after a process change increased load on the asset. The corrective action adjusts the interval and adds a condition check at each service visit. Two cycles later, no recurrence. That's corrective action.
On the other hand, imagine work order history shows three similar pumps have each needed unscheduled bearing replacements in the past year, but the fourth hasn't failed yet. The team inspects it, replaces the bearing on condition, and updates the PM interval for all four before anything breaks. No failure event triggered the work. That's preventive action.
CAPA in Maintenance Operations
In maintenance operations, CAPA is triggered by repeated equipment failures, failed audits, deviation reports from production, and near-miss reports that surface latent conditions before they produce a failure.
Asset history functions as CAPA evidence. When an investigator asks why an asset failed, the answer is in the maintenance record: How many times has this failure mode appeared in the last 24 months? Was the most recent PM completed on schedule? Were the correct parts and specifications used?
A CMMS linking every work order to the asset makes that question answerable in minutes. Without it, the question requires days of reconstruction and produces a record auditors will probe for gaps.
PM compliance rate is the upstream variable the corrective action queue runs on. When scheduled maintenance holds, developing conditions are caught before they produce failures.
For facilities in FDA-regulated environments, pharma, medical device manufacturing, and food processing, CAPA is a compliance requirement. The maintenance management system is part of the quality system, and its records are auditable. Gaps in those records carry the same regulatory exposure as gaps in the CAPA documentation itself.
Implementing CAPA in Your Maintenance Program
A functioning CAPA program requires connected documentation infrastructure. Without it, root cause analysis starts with incomplete evidence, verification steps are dropped, and CAPA records are reconstructed under audit pressure from whatever files can be located.


Step 1: Establish a formal intake process
Every failure event, deviation, or risk signal needs a documented starting point with enough specificity to support investigation. In a CMMS, this is the work order or deviation report.
An email or verbal report isn’t a documented starting point. It’s a record that doesn’t exist when the CAPA is reviewed.
Step 2: Connect work order history to the investigation
Root cause analysis requires pattern recognition across time. A CMMS linking every work order to the asset record makes it possible to determine whether a failure has occurred before, how frequently, what actions were taken, and whether those actions held.
Step 3: Document the root cause determination separately from the immediate repair
The repair record and the CAPA record serve different purposes and shouldn’t be combined. The repair record documents what was done to restore function. The CAPA record documents why the failure occurred and what systemic change was made to prevent recurrence.
Step 4: Build the verification check into the CAPA workflow before closing
Define the effectiveness criterion at the time the corrective action is written, not when the CAPA is reviewed for closure. Then, set a follow-up date. When that time arrives, pull the asset's work order history and apply the pre-defined criterion.
Step 5: Track open CAPAs by age and status
Managers need visibility into how many CAPAs are open, for how long, and which assets they’re tied to. CAPAs aging past their target closure date without escalation accumulate regulatory exposure.
UpKeep connects each step of this workflow in a single audit-ready system. A failure event opens a work order. It then links to the asset's full maintenance history so pattern recognition is part of the investigation from the start rather than a separate reconstruction effort.
The root cause determination and the corrective action are documented in the record attached to the asset profile. When a regulatory inspector requests documentation on a specific asset, the triggering work order, the action plan, and the subsequent PM history are all accessible from a single asset view.
CAPA Metrics Worth Tracking
The following five metrics give a complete read on whether your CAPA program is functioning or starting to break down.
|
Metric |
What It Measures |
Warning Signal |
|---|---|---|
CAPA Cycle Time |
Average days from CAPA opened to CAPA closed |
Extended timelines indicate bottlenecks in root cause investigation or action plan approval. Track corrective and preventive actions separately; preventive actions typically have longer, more ambiguous investigation timelines |
Recurrence Rate |
Percentage of closed CAPAs where the same failure mode reappears within a defined window |
High recurrence signals root cause investigations are stopping at the symptom level. The program is generating repeated work on the same assets |
|
Effectiveness Verification Completion Rate |
Percentage of closed CAPAs with a documented effectiveness check on file |
Should be at or near 100% in a functioning program. Anything below that requires a structural control in the intake and closure workflow, not a reminder |
CAPA Backlog Age |
Number of open CAPAs past their target closure date |
Aging CAPAs accumulate regulatory exposure and signal a program that can’t close actions within its own timelines. In regulated environments, aged open CAPAs are a finding before an inspector reviews investigation quality |
PM Compliance Rate |
Percentage of scheduled PMs completed on time (tracked as a leading indicator) |
Consistent PM completion reduces the failure rate feeding the CAPA queue. Compliance gaps precede failure spikes by weeks to months; the CAPA program absorbs the downstream work |
From Reactive Documentation to a Defensible Quality Record
Without connected documentation, an investigation starts with whatever records someone can locate under pressure. The root cause stays out of reach. The verification check gets dropped because nothing enforces it. Each closed CAPA without one becomes evidence in the next audit.
When the program works, failure events carry timestamped work orders tied to the full asset history. The investigation starts with the complete picture, not a reconstruction. The root cause determination is documented apart from the repair, and the verification criterion is set before the action closes.
UpKeep provides the infrastructure connecting failure events to work orders, work orders to asset history, and asset history to the corrective action record. When the maintenance and the quality documentation systems are the same, CAPA records are built as work occurs, not reconstructed when an auditor asks for them.
Curious how UpKeep handles this in practice? Reach out and we'll show you.
FAQs About Corrective and Preventive Action
What is the difference between corrective action and preventive action?
Corrective action deals with a problem that’s already occurred. Preventive action deals with a potential problem that hasn't happened yet. The trigger determines which one applies: Actual failure calls for corrective action, while a data trend or risk signal calls for preventive.
What is the difference between a correction and a corrective action?
A correction fixes the immediate problem: replace the part, restore function, etc.. A corrective action finds out why the failure happened and changes the condition that caused it. Without that second step, the root cause stays in place and the failure comes back eventually.
How do you verify that a corrective action was effective?
Most programs don't, which is the gap. Verification requires a defined criterion set before the action closes, not after, and a follow-up date that someone is actually responsible for. For equipment failures, two complete PM cycles without recurrence is a practical standard. Without a system enforcing the follow-up, the check gets dropped and the CAPA closes incomplete.
What triggers a CAPA in a maintenance context?
Corrective action triggers are visible: repeated failures, failed audits, or deviation reports. Preventive action triggers are harder, taking the form of work order trends, risk assessments, or audit observations about PM gaps that haven't produced a failure yet. The difficulty is that preventive triggers only surface if someone is actively looking at the data. In a reactive operation, that rarely happens without a system making it visible.
What does an FDA inspector look for in a CAPA program?
Consistent documentation of both corrective and preventive action as separate processes, per 21 CFR 820.100. The most common findings are: CAPAs closed without a verification check, investigations that stop at the proximate cause, corrective and preventive actions treated as one process, and records that can't be tied back to the underlying asset data.
4,000+ COMPANIES RELY ON ASSET OPERATIONS MANAGEMENT
Leading the Way to a Better Future for Maintenance and Reliability
Your asset and equipment data doesn't belong in a silo. UpKeep makes it simple to see where everything stands, all in one place. That means less guesswork and more time to focus on what matters.









