Blog Post
Facility Maintenance in Pharmaceutical Manufacturing: The GMP Compliance Guide
Keep pharmaceutical facilities FDA-compliant with structured PM programs, validated cleaning procedures, and CMMS-backed documentation for inspection readiness.



Key Takeaways
Facility maintenance for pharmaceutical manufacturing is a compliance function, not a support service. Under FDA 21 CFR Part 211, buildings and facilities must be maintained in a good state of repair, and unplanned equipment failures carry audit exposure that reactive programs can’t absorb.
Maintenance records are the deliverable. FDA regulations require cleaning, inspection, and sanitizing records to be kept chronologically with dated signatures. A CMMS that generates these records automatically from completed work orders closes the gap between work performed and work provable.
Calibration gaps turn instrument overdue dates into data integrity problems. When sensors, scales, or autoclaves fall outside their calibration window, every measurement and batch record produced during that period is called into question. A risk-tiered schedule with overdue alerts prevents this from becoming a retrospective problem.
Paper-based maintenance programs frequently fail the FDA’s documentation standard at scale. Investigators expect records that are accurate, traceable, attributable, and contemporaneous. Across a multi-shift, multi-technician operation, paper rarely delivers all four consistently.
In pharmaceutical manufacturing, a maintenance failure is a regulatory event. When critical equipment fails without a documented preventive maintenance history, the repair is the least urgent concern. The immediate question is whether every batch produced on that equipment since the last confirmed service event can still be released, or whether those batches must be quarantined, investigated, and potentially recalled. That determination must be made before production resumes, not after.
The FDA inspects drug manufacturers on a risk-based schedule. Facilities with compliance histories, product risk profiles, or prior observations are prioritized, and maintenance records are among the first aspects an investigator requests. A facility with a reactive program that services equipment when it fails rather than on a documented schedule generates record gaps that lead to the dreaded Form 483 observations. In confirmed cases, those observations escalate to warning letters, consent decrees, and import alerts that can shut down production lines entirely. The FDA’s authority to act on 21 CFR Part 211 violations is broad, and it wields it harshly.
Maintenance isn’t a support function in pharmaceutical manufacturing but a production control system. The FDA treats it that way, and so do the manufacturers that stay out of enforcement action.
What Is Facility Maintenance in Pharmaceutical Manufacturing?
Pharmaceutical facility maintenance is the structured management of buildings, equipment, utilities, and environmental systems to sustain current Good Manufacturing Practice (cGMP) compliance and product integrity. Compared to general industrial maintenance, it carries greater consequences for failure. In a regulated manufacturing environment, a lapsed calibration or an unvalidated cleaning procedure poses a product-quality risk and a potential patient-safety exposure.
The regulatory framework that governs this work is cGMP. The “current” isn’t incidental: It means manufacturers are required to use up-to-date technologies, systems, and controls, not simply comply with a fixed standard written at a point in time. The FDA updates its expectations as science and manufacturing practice evolve, and a maintenance program built around outdated methods risks falling short of current expectations, even if the same approach was accepted five years ago.
The regulatory scope covers four domains: buildings and facilities, equipment, utilities, and documentation. Each carries specific requirements under FDA cGMP regulations and, for manufacturers of Active Pharmaceutical Ingredients (APIs), which are the compounds responsible for a drug’s therapeutic effect, the ICH Q7 guidance published by the International Council for Harmonisation, the body that aligns pharmaceutical regulatory requirements across the US, EU, and Japan. In pharmaceutical manufacturing, informal is non-compliant.
The Regulatory Framework: What FDA and GMP Actually Require
Buildings and facilities: Under §211.42, pharmaceutical buildings must be of suitable size, construction, and location to facilitate cleaning, maintenance, and proper operations. §211.58 states that any building used in drug manufacturing must be maintained in a good state of repair. These are the baseline below which an FDA investigator will cite a violation.
Equipment cleaning and maintenance: §211.67(a) requires equipment and utensils to be cleaned, maintained, and, where necessary, sanitized or sterilized at appropriate intervals to prevent malfunctions or contamination that would alter the safety, identity, strength, quality, or purity of the drug product. §211.67(b) requires written Standard Operating Procedures (SOPs) establishing responsibility for cleaning and maintenance, schedules, methods, and pre-use inspection. The FDA’s Q&A guidance on cGMP requirements for equipment reinforces that these procedures must be well documented and validated, not described in a general SOP that technicians interpret in the field.
Documentation: §211.182 requires a cleaning and use log for each piece of major equipment. Entries must be chronological, dated, and signed by the person performing the work. Where dedicated equipment is used, these records become part of the batch record, meaning a gap in the maintenance log is a gap in the batch record. For shared equipment, a maintenance gap is equally problematic because it undermines the evidentiary chain that production conditions were controlled.
Utilities for API manufacturers: ICH Q7 Section IV.B requires that all utilities capable of impacting product quality, such as steam, gases, compressed air, and HVAC (Heating, Ventilation, and Air Conditioning), be qualified and monitored with action limits. ICH Q7 uses guidance language, but FDA investigators routinely apply these expectations during API facility inspections. In practice, they function as the benchmark against which facilities are measured. A facility that services its HVAC on a calendar schedule without qualification documentation is operating outside what inspectors expect to see.
The Four Maintenance Domains: Regulatory Anchors at a Glance
|
Maintenance Domain |
Regulatory Anchor |
Scope & Key Requirements |
|---|---|---|
|
Equipment PM & Calibration |
21 CFR §211.67(a)(b)(c)§211.182 |
Risk-tiered PM schedules for all critical assets; calibration program with defined intervals, overdue alerts, and certificates linked to asset records |
|
HVAC & Cleanroom Systems |
ICH Q7 Section IV.B21 CFR §211.46 |
Pressure differentials, particle counts, HEPA filter integrity; IQ/OQ/PQ documentation required; re-qualification after significant interventions |
|
Utilities |
ICH Q7 Section IV.B21 CFR §211.65 |
Purified water, compressed air, and steam; each validated and monitored with action and alert limits; failure can contaminate entire production runs |
|
Facility Fabric |
21 CFR §211.56§211.58 |
Smooth, coved, cleanable surfaces; drain maintenance and pest control logs; cracks and deteriorating seals are documented deficiencies, not housekeeping issues |
Equipment PM and Calibration
Critical manufacturing equipment requires a predictive maintenance (PM) schedule built around criticality (how directly an asset affects product quality). High-criticality assets, including tablet presses, autoclaves, mixing vessels, or centrifuges, carry tighter intervals and full documentation of every service event. Calibration runs as a parallel discipline: Sensors, scales, temperature monitors, and pressure gauges must be calibrated on a defined schedule, with certificates stored against the asset record.
When a calibration lapses and a batch is produced during the overdue window, every measurement generated in that period is suspect. Correcting this retrospectively requires a deviation investigation, a risk assessment, and potentially a batch disposition decision. A functioning overdue alert makes all of this avoidable.
HVAC and cleanroom systems
HVAC in a pharmaceutical facility is a quality control system, not building infrastructure. Pressure differentials between classified and unclassified spaces prevent contamination migration, so particle counts, temperature, and humidity must remain within validated limits. HEPA filter integrity requires scheduled testing, not visual inspection.
Qualification documentation, such as Installation Qualification (IQ), Operational Qualification (OQ), and Performance Qualification (PQ) must be on file and current. For significant interventions, including major repairs, filter replacements or structural changes, requalification is typically expected, with the scope determined by the nature and risk of the change. A cleanroom returned to service after a significant intervention without any requalification assessment is an audit finding waiting to be discovered.
Utilities: purified water, compressed air, steam
Each of these utilities can introduce contamination directly into the manufacturing process if not properly maintained and monitored. Purified water systems require validated monitoring with action and alert limits. Compressed air in contact with product or product-contact surfaces requires oil-free systems and regular purity testing. Steam used in sterilization requires pressure and temperature validation. Utility failure at the point of use may not be immediately visible, which is why monitoring with action limits, rather than periodic inspection alone, is the required control mechanism.
Facility fabric: surfaces, drains, and pest control
cGMP requires pharmaceutical facilities to be maintained in a clean and sanitary condition, free of infestation. Surfaces must be smooth, non-porous, coved at floor-wall junctions, and cleanable without harboring microbial growth. Cracks, peeling paint, and deteriorating seals are documentable deficiencies.
A 2025 FDA warning letter cited Chem-Tech Ltd. for failing to maintain the facility in a good state of repair, with the investigator observing visible contamination on equipment that had not been cleaned between production runs. Drain maintenance and pest control logs are part of the facility’s audit record, not optional background documentation.
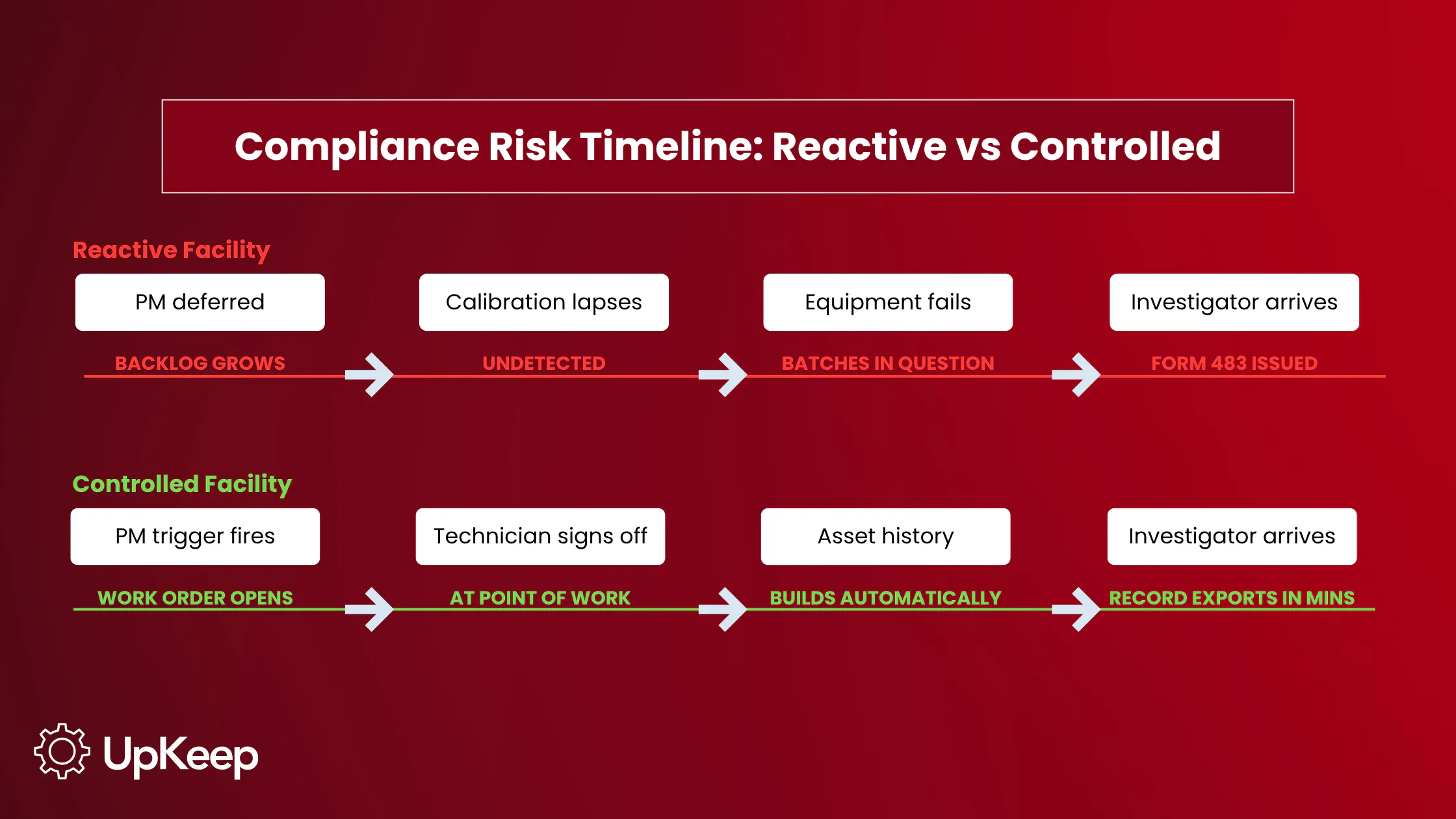
Reactive Program vs. GMP-Compliant Program


|
Area |
Reactive / Break-Fix |
GMP-Compliant Program |
|---|---|---|
|
PM scheduling |
Serviced after failure; no defined intervals or written procedures |
Risk-tiered schedule with documented intervals; every deviation triggers an investigation |
|
Documentation |
Paper logs completed after the fact; timestamps and signatures inconsistent |
CMMS captures electronic signatures at task completion; timestamp set by system, not technician |
|
Calibration |
Tracked by memory or spreadsheet; overdue instruments go undetected |
Risk-based intervals with automated overdue alerts; certificates stored in the asset record |
|
HVAC management |
Serviced on calendar schedule; no IQ/OQ/PQ documentation on file |
Qualification documentation current; re-qualification required after significant interventions |
|
Audit readiness |
Records assembled under pressure; gaps surface during inspection |
Asset history complete and exportable on demand; no reconstruction required |
Maintenance Documentation: What the FDA Expects
The FDA’s documentation standard goes beyond record-keeping. Every maintenance event must be documented at the time it occurs by the person who performed it and on the specific asset it covers. The standard is clear: Records must be accurate, attributable, contemporaneous, and legible—what the FDA calls the ALCOA principles.
Records completed well after the work is done (or signed at shift’s end rather than at task completion) weaken your “up to date” claims and create the kind of documentation pattern that draws investigator scrutiny, regardless of whether the underlying work was performed correctly.
In practice, every service event generates a record covering who performed the work, what was done, when, on which asset, and the asset’s condition before and after. Written SOPs must be established for each piece of equipment and followed consistently. Equipment must be inspected for cleanliness immediately before use, and that inspection must be recorded. These are the conditions under which maintenance work is considered compliant.
Paper-based programs frequently fail to meet this standard not because of intent but consistency. Across a multi-shift operation with rotating technicians, paper logs accumulate the kind of gaps that undermine compliance: entries completed hours after the work, signatures added at shift’s end rather than at task completion, cleaning records that describe the procedure rather than confirming it was performed on that asset, at that time, by that person.
When an investigator requests documentation for a specific asset across six months, a paper-based program produces a reconstruction. That reconstruction is exactly what the contemporaneous standard prohibits.
A CMMS (computerized maintenance management system) closes that gap structurally. Work orders open from PM triggers and can’t be marked complete without a technician’s signature at the point of work. The timestamp is set by the system, not entered manually, and the asset history builds record by record as work is performed, not assembled under audit pressure. When electronic records are used this way, 21 CFR Part 11, the FDA’s regulation governing electronic records and signatures, requires audit trails, access controls, and data integrity equivalent to paper. A properly configured CMMS satisfies that standard as a product of normal operations.
KPIs for Pharmaceutical Facility Maintenance
Tracking the right metrics separates a maintenance program that feels compliant from one that can prove it. The following six KPIs give operations and quality teams the earliest possible signal when the program is drifting, before it becomes a deviation that leads to a Form 483 observation. The last metric, Corrective and Preventive Action (CAPA) closure time, is particularly watched during inspections, as open CAPAs signal that the quality system is identifying problems but not resolving them.
|
KPI |
What It Measures |
Why It Matters for GMP Compliance |
|---|---|---|
|
PM Completion Rate |
% of scheduled PM tasks completed on time |
Many pharmaceutical programs target 90%+. Consistent shortfalls signal deferred maintenance accumulating, which surfaces as emergency repairs and potential record gaps within 30–60 days. |
|
Calibration Overdue Rate |
# of instruments outside their calibration window |
Any critical instrument outside its window requires an immediate deviation investigation. Every measurement produced during the overdue period is suspect until assessed and documented. |
|
Corrective-to-Preventive Work Order Ratio |
Unplanned repairs vs. scheduled maintenance |
A rising ratio means the PM program is losing ground to break-fix. High corrective volume is both an operational cost signal and a leading indicator of audit exposure. |
|
Equipment Downtime % |
Time assets are unavailable due to maintenance |
Tracks the operational cost of the maintenance program. Unplanned downtime on critical equipment opens batch record integrity questions that require documented risk assessment. |
|
CAPA Closure Time |
Avg. days from deviation to corrective action closure |
Extended CAPA timelines increase regulatory exposure. Open CAPAs are reviewed during FDA inspections and signal that the quality system isn’t closing its own gaps. |
|
Work Order Backlog Age |
Average age of open work orders |
A growing backlog on critical equipment is an operational and compliance risk. Investigators treat backlog age as evidence of whether the PM program is functioning or stalled. |
How to Build a GMP-Compliant Facility Maintenance Program
1. Complete an asset inventory and assign criticality classifications: Every piece of equipment, utility system, and facility component should be inventoried and classified by its direct impact on product quality. The classification framework typically runs three tiers: direct product contact and quality-critical equipment at the top, indirect contact equipment in the middle, and non-production infrastructure at the base.
Each tier carries different PM intervals, documentation requirements, and deviation investigation depth. Without this map, maintenance resources spread evenly across assets that carry uneven compliance risk, and the highest-risk equipment receives the same attention as a utility room HVAC unit. Classification is the decision that makes everything downstream defensible.
2. Build PM schedules by risk tier: High-criticality assets require manufacturer-recommended service intervals as a floor, not a ceiling. Actual intervals should be informed by failure history, production volume, environmental conditions, and the consequences of an unplanned failure on in-process or released batches. For equipment with limited failure history, start at manufacturer recommendations and tighten based on observed wear patterns over the first 12–18 months of operation.
Every PM task must specify what’s inspected, what’s replaced, what constitutes an acceptable outcome, and what triggers a deviation when the outcome falls outside specification. A PM that completes without a documented pass/fail outcome is not a compliant PM.
3. Develop and version-control SOPs for cleaning and maintenance: Written procedures are a regulatory requirement. Each SOP must assign responsibility, specify the method, identify the materials used, and include a pre-use inspection step. SOPs must be version-controlled, accessible to technicians at the point of work, and updated whenever equipment, materials, or procedures change.
A change to a cleaning agent without a corresponding SOP revision is a deviation, regardless of whether the cleaning outcome was acceptable. Version control allows an investigator to confirm the procedure followed on a given date matches what was in effect on that date.
4. Establish a calibration program with risk-based intervals and automated alerts: Group instruments by criticality and assign calibration intervals accordingly. Critical instruments whose measurements directly inform batch release decisions require tighter intervals and immediate escalation protocols when overdue.
An automated alert before calibration is due eliminates the manual tracking that allows overdue instruments to slip through in high-volume operations. Calibration certificates should be stored in the asset record so that any inquiry about a specific batch can be answered by pulling the asset history rather than by searching paper files.
5. Select a documentation system that meets the FDA’s contemporaneous record standard: For facilities beyond the smallest scale, paper can’t reliably satisfy the FDA’s accuracy, traceability, and contemporaneity requirements.
A CMMS that generates work orders from PM triggers, captures electronic signatures at task completion, and maintains a timestamped audit trail for every asset can produce the documentation an FDA investigator expects. UpKeep centralizes PM scheduling, work order management, and asset history on a single platform, with mobile access that puts documentation at the point of work.
6. Train all maintenance personnel on GMP principles, SOPs, and documentation requirements: A technically skilled technician who doesn’t understand why documentation accuracy matters is a compliance risk. Training must cover cGMP principles, the SOPs governing each piece of equipment they service, documentation standards, contamination control practices, and the change control process for reporting equipment modifications.
Training records are themselves a regulatory requirement and must be documented, dated, and current. An untrained technician performing compliant work leaves no proof that the work met the standard.
Expand Facility Maintenance Into a Compliance System
A pharmaceutical facility running on reactive maintenance doesn't fail visibly. It accumulates. Equipment is serviced after it breaks. Calibration slips past its window while production continues. Cleaning records are filled in at the end of shifts by whoever is around. HVAC runs on a calendar because nobody qualified it. None of these small faults looks like a crisis until an investigator asks for six months of asset history, and the program has to reconstruct it from memory.
That reconstruction is the problem. The maintenance gaps themselves are fixable, but proving the work was done, when, and by whom is a greater obstacle. A controlled program solves that at the source: PM runs on schedule, calibration stays current, and every service event is logged at the point of work by the technician who performed it. When documentation is built into how the work gets done rather than added afterward, there's nothing to reconstruct.
UpKeep makes that the default. Work orders open automatically from PM triggers, and technicians sign off on mobile at the point of work, so the timestamp is the system's, not whoever completes the paperwork. Asset history is exportable in minutes, meaning an investigator who arrives unannounced receives the same record as one who schedules a visit three weeks out.
FAQ
What does FDA 21 CFR Part 211 require for pharmaceutical facility maintenance?
21 CFR Part 211 Subpart C requires facilities to be maintained in a good state of repair under §211.58. Subpart D requires equipment to be cleaned and maintained at appropriate intervals, with written procedures governing those activities. Records of maintenance, cleaning, sanitizing, and inspection must be kept chronologically with dated signatures under §211.67(c) and §211.182.
How is pharmaceutical facility maintenance different from standard industrial maintenance?
The technical work is often similar. The difference is consequence and documentation. In pharmaceutical manufacturing, a maintenance gap creates product quality risk and regulatory exposure. In a standard industrial setting, the same gap creates a repair backlog. Every maintenance event in pharma must be documented in a way that’s attributable, accurate, and contemporaneous. In most other industries, that level of documentation is optional.
What maintenance records does the FDA expect to see during an inspection?
Investigators typically request equipment cleaning and use logs, written SOPs for cleaning and maintenance, calibration records linked to each asset, deviation reports for any departure from procedure, and documentation of corrective actions. Records should be timestamped, signed by the performing technician, and retrievable by asset and date range without manual reconstruction.
What types of equipment require calibration in a pharmaceutical facility?
Any instrument whose measurements influence product quality decisions requires a calibration program. This includes temperature sensors, pressure gauges, scales and balances, pH meters, autoclaves, and environmental monitoring instruments in classified spaces. Calibration frequency is set by criticality, manufacturer specifications, and historical performance, not by a single universal interval.
How does a CMMS support GMP compliance in pharmaceutical manufacturing?
A CMMS automates PM scheduling so tasks fire on the defined interval without manual tracking. It generates work orders that technicians complete and sign electronically, creating timestamped records that satisfy the FDA’s contemporaneous documentation requirements. Calibration due dates are tracked at the asset level with alerts before the window closes, and asset history and maintenance logs export on demand. Under 21 CFR Part 11, a CMMS with audit trail and electronic signature capabilities produces electronic records that meet the same reliability and traceability standard as paper.
What are the most common maintenance-related FDA warning letter violations?
Recurring citations include failure to maintain the facility in a good state of repair, failure to establish or follow written cleaning and maintenance procedures, absence of a preventive maintenance program, and inadequate or incomplete maintenance records. Industry analysis of 2025 FDA warning letters by Leucine found that quality system violations—a category that includes maintenance program deficiencies—account for over 30% of all citations.
4,000+ COMPANIES RELY ON ASSET OPERATIONS MANAGEMENT
Leading the Way to a Better Future for Maintenance and Reliability
Your asset and equipment data doesn't belong in a silo. UpKeep makes it simple to see where everything stands, all in one place. That means less guesswork and more time to focus on what matters.









